
(2S,3S,11βS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11β-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-(4S)-fluoromethyl-pyrrolidin-2-one Dihydrochloride
(2S,3S,11bS)-1-(2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4(S)-fluoromethyl-pyrrolidin-2-one
813452-14-1 (di-HCl)
916069-91-5 (mono-HCl)
Roche…….innovator
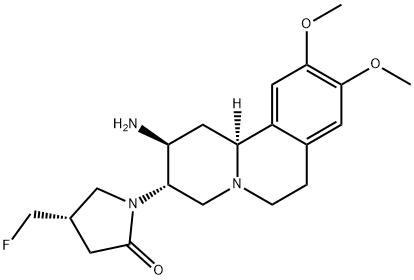
CARMEGLIPTIN
813452-18-5
(2S,3S,11βS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11β-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-(4S)-fluoromethyl-pyrrolidin-2-one
(S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinolin-3-yl)-4-(fluoromethyl)pyrrolidin-2-one
| (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinolin-3-yl)-4-(fluoromethyl)pyrrolidin-2-one | |
| (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-2,3,4,6,7,11b-hexahydro-1H-pyrido[2,1-a]isoquinolin-3-yl)-4-(fluoromethyl)pyrrolidin-2-one | |
| 分子式: | C20H28FN3O3 |
| 分子量: | 377 |
813452-18-5, Carmegliptin, R-1579;carmegliptin, Carmegliptin (USAN/INN), SureCN419289, UNII-9Z723VGH7J, CHEMBL591118, CHEBI:699093, Ro-4876904, D08631, R-1579, B1Q
Type 2 diabetes is a chronic, progressive metabolic disease, affecting about 4% of the world population. The main goal of the management of type 2 diabetes is to achieve glycemic control as close to the nondiabetic range as practicable, in order to reduce the risk of late-stage complications.However, the therapeutic effect provided by existing medications is often not sustainable, since the multi-organ defects responsible for the disease are only insufficiently addressed.
Dipeptidyl peptidase-IV (DPP-IV) inhibitors have emerged as a new therapeutic option to treat type 2 diabetes.
Their rapid rise in popularity is due to the favourable safety profile (no hypoglycemia, no weight gain, no gastrointestinal problems—typical side effects associated with established anti-diabetic agents). DPP-IV is a ubiquitous serine protease, the inhibition of which prevents the degradation of glucagon-like peptide 1 (GLP-1). The resulting higher levels of GLP-1 have a beneficial impact on major players involved in the pathogenesis of type 2 diabetes: β-cells, liver, α-cells, gut, and brain.
Long-term studies with DPP-IV inhibitors in patients are underway in order to confirm the safety and sustainability of these effects, and, in particular, their ability to prevent the progressive loss of β-cell function.
SYNTHESIS

aReagents and conditions: a) HCO2Me, Δ; b) POCl3, MeCN; c) HO2CCH2CO2Et, neat, 120 °C; d) ethyl acrylate, neat; e) t-BuOK, neat (5 steps); f) NH4OAc, MeOH; g) NaBH4, TFA, THF; h) Boc2O, CH2Cl2; i) KOH, aq THF; j) DPPA, Et3N, TMSCH2CH2OH, PhMe, 80 °C; k) Et4NF, MeCN; l) chiral HPLC; m) Et3N, CH2Cl2; n) NaH, DMF; o) HCl, dioxane; p) HCl, 2-PrOH.

Reagents and conditions: (a) NH4OAc, MeOH, rt, 95%; (b) NaBH4, TFA, THF, 0 °C; (c) Boc2O, CH2Cl2, 83% over 2 steps; (d) KOH, aq THF, rt; (e) DPPA, Et3N, 2-(trimethylsilyl)ethanol, toluene, 80 °C; (f) Et4NF, CH3CN, 50 °C, 56% over 3 steps; (g) Et3N, CH2Cl2, (h) NaH, cat. NaI, DMF; (i) HCl, 1,4-dioxane.
Carmegliptin (2.70) is an anti-diabetes drug which is currently in late stage clinical trials. It represents a further structural advancement from the other existing marketed drugs in this class, sitagliptin (2.71, Januvia) and vildagliptin (2.72, Zomelis, Figure 7). These compounds are all members of the dipeptidyl peptidase 4 class (DPP-4), a transmembrane protein that is responsible for the degradation of incretins; hormones which up-regulate the concentration of insulin excreted in a cell. As DPP-4 specifically cleaves at proline residues, it is unsurprising that the members of this drug class exhibit an embedded pyrrolidine ring (or mimic) and additional decoration (a nitrile or fluorinated alkyl substituent is present in order to reach into a local lipophilic pocket). One specific structural liability of the 2-cyano-N-acylpyrrolidinyl motif (2.73) is its inherent susceptibility towards diketopiperazine formation (2.74, Scheme 29) [80], however, one way to inhibit this transformation is to position a bulky substituent on the secondary amine nucleophile as is the case in vildagliptine (2.72).
![[1860-5397-9-265-7]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-7.png?max-width=550&background=EEEEEE)
![[1860-5397-9-265-i29]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i29.png?max-width=550&background=EEEEEE)
A single crystal X-ray structure of carmegliptin bound in the human DPP-4 active site has been published indicating how the fluoromethylpyrrolidone moiety extends into an adjacent lipophilic pocket [81]. Additional binding is provided by π–π interaction between the aromatic substructure and an adjacent phenylalanine residue as well as through several H-bonds facilitated by the adjacent polar substituents (Figure 8).
![[1860-5397-9-265-8]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-8.png?max-width=550&background=EEEEEE)
The reported synthesis of carmegliptin enlists a Bischler-Napieralski reaction utilising the primary amine 2.76 and methyl formate to yield the initial dihydroquinoline 2.77 as its HCl salt (Scheme 30) [82]. This compound was next treated with 3-oxoglutaric acid mono ethyl ester (2.78) in the presence of sodium acetate. Decarboxylation then yields the resulting aminoester 2.79 which was progressed through an intramolecular Mannich-type transformation using aqueous formaldehyde to allow isolation of enaminoester 2.80 after treatment of the intermediate with ammonium acetate in methanol.
The next step involves a very efficient crystallisation-induced dynamic resolution of the racemic material using the non-natural (S,S)-dibenzoyl-D-tartaric acid ((+)-DBTA). It is described that the desired (S)-enantiomer of compound 2.81 can be isolated in greater than 99% ee and 93% overall yield. This approach is certainly superior to the original separation of the two enantiomers (at the stage of the final product) by preparative chiral HPLC that was used in the discovery route (albeit it should be noted that both enantiomers were required for physiological profiling at the discovery stage).
Next, a 1,2-syndiastereoselective reduction of enaminoester 2.81 occurs with high diastereocontrol imposed by the convexed presentation of the substrate for the formal conjugate addition and subsequent protonation steps. This is followed by Boc-protection and interconversion of the ethyl ester to its amide derivative 2.82 in 80% overall yield for this telescoped process. The primary amide in 2.82 was then oxidised via a modern variant of the classical Hoffmann rearrangement using phenyliodine diacetate (PIDA).
Following extensive investigation it was found that slowly adding this reagent in a mixture of acetonitrile/water to a suspension of amide 2.82 and KOH gave clean conversion to the amine product in high yield. This new procedure was also readily scalable offering a cleaner, safer and more reliable transformation when compared to other related rearrangement reactions. During a further telescoped procedure amine 2.83 was treated with lactone 2.84 to regenerate the corresponding lactam after mesylate formation. Finally, removal of the Boc-group with aqueous hydrochloric acid furnished carmegliptin as its HCl salt.
![[1860-5397-9-265-i30]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i30.png?max-width=550&background=EEEEEE)
- Peters, J.-U. Curr. Top. Med. Chem. 2007, 7, 579–595……………..80
- Mattei, P.; Boehringer, M.; Di Gorgio, P.; Fischer, H.; Hennig, M.; Huwyler, J.; Koçer, B.; Kuhn, B.; Loeffler, B. M.; MacDonald, A.; Narquizian, R.; Rauber, E.; Sebokova, E.; Sprecher, U.Bioorg. Med. Chem. Lett. 2010, 20, 1109–1113. doi:10.1016/j.bmcl.2009.12.024………..81
- Albrecht, S.; Adam, J.-M.; Bromberger, U.; Diodone, R.; Fettes, A.; Fischer, R.; Goeckel, V.; Hildbrand, S.; Moine, G.; Weber, M. Org. Process Res. Dev. 2011, 15, 503–514. doi:10.1021/op2000207……….82
………………………………………………………………………………………………………………..
Org. Process Res. Dev. 2011, 15, 503–514. doi:10.1021/op2000207
http://pubs.acs.org/doi/full/10.1021/op2000207

A short and high-yielding synthesis of carmegliptin (1) suitable for large-scale production is reported. The tricyclic core was assembled efficiently by a decarboxylative Mannich addition−Mannich cyclization sequence. Subsequent crystallization-induced dynamic resolution of enamine 7 using (S,S)-dibenzoyltartaric acid was followed by diastereoselective enamine reduction to give the fully functionalized tricyclic core with its three stereogenic centers. The C-3 nitrogen was introduced by Hofmann rearrangement of amide 28, and the resulting amine 10was coupled with (S)-fluoromethyl lactone 31. Following cyclization to lactam 13 and amine deprotection, 1 was obtained in 27−31% overall yield with six isolated intermediates.
Preparation of (2S,3S,11βS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11β-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-(4S)-fluoromethyl-pyrrolidin-2-one Dihydrochloride (1) CARMEGLIPTIN
Bioorg Med Chem Lett 2010, 20(3): 1109
-
Discovery of carmegliptin: A potent and long-acting dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes
- Pages 1109-1113
- Patrizio Mattei, Markus Boehringer, Patrick Di Giorgio, Holger Fischer, Michael Hennig, Joerg Huwyler, Buelent Koçer, Bernd Kuhn, Bernd M. Loeffler, Alexander MacDonald, Robert Narquizian, Etienne Rauber, Elena Sebokova, Urs Sprecher
-

![Full-size image (16 K)]() Scheme 3.
Scheme 3.Reagents and conditions: (a) preparative HPLC (Chiralpak® AD column), heptane/2-propanol 85:15, 37% (b) BH3.Me2S, THF, 0 °C; (c) (MeOCH2CH2)2NSF3, CH2Cl2, 67% (2 steps); (d), SOCl2, ZnCl2, 80 °C, 72 h, 61%; (e) Et3N, CH2Cl2; (f) NaH, DMF, 56% (2 steps); (g) HCl, 1,4-dioxane, 91%; (h) HCl, 2-propanol, 86%.


The most preferred product is (2S,3S,11bS)-2-tert.-Butoxycarbonylamino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H pyrido[2,1-a]isoquinoline-3-carboxylic acid amide having the following structure:

It has been found that during the amidation of the ester epimerization takes place at position 3 and thus the 3R-epimer of the formula IVb is transformed to a larger extent in the 3S-epimer of formula V.

e) Preparation of (2S,3S,11bS)-1-(2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4(S)-fluoromethyl-pyrrolidin-2-one Dihydrochloride
A 2.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 619 g (1.30 mol) of (2S,3S,11bS)-3-((4S)-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester, 4.2 L isopropanol and 62 mL water and the suspension was heated to 40-45° C. In a second vessel, 1.98 L isopropanol was cooled to 0° C. and 461 mL (6.50 mol) acetyl chloride was added during 35 min, maintaining the temperature at 0-7° C. After completed addition, the mixture was allowed to reach ca. 15° C. and was then slowly added to the first vessel during 1.5 h. After completed addition the mixture was stirred for 18 h at 40-45° C., whereas crystallization started after 1 h. The white suspension was cooled to 20° C. during 2 h, stirred at that temperature for 1.5 h and filtered. The crystals were washed portionwise with 1.1 L isopropanol and dried for 72 h at 45° C./20 mbar, to give 583 g of the product as white crystals (100% yield; assay: 99.0%).







MS: m/e 496 (M+H)+, 437.
d) Preparation of (2S,3S,11bS)-3-((4S)-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl esterA 1.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel, a cooling bath and a nitrogen inlet was charged with 28 g (56.5 mmol) of (2S,3S,11bS)-3-(3-fluoromethyl-4-hydroxy-butyrylamino) -9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester and 750 mL THF. The mixture was cooled to 0° C. and a solution of 6.17 mL (79 mmol) methanesulfonic acid in 42 mL THF was added during 10 min, maintaining the temperature at 0-5° C. At 0° C. a solution of 12.6 mL (90.2 mmol) triethylamine in 42 mL THF was added during 15 min. The resulting suspension was stirred for 80 min at 0-5° C., whereas it became gradually thicker. Then 141 mL (141 mmol) 1 M lithium-bis(trimethylsilyl)amide were added to the mixture during 15 min, whereas the suspension dissolved. The solution was allowed to reach RT during 60 min under stirring. 500 mL water was added without cooling, the mixture was extracted and the aqueous phase was subsequently extracted with 500 mL and 250 mL dichloromethane. The organic layers were each washed with 300 mL half saturated brine, combined and evaporated on a rotatory evaporator. The resulting foam was dissolved in 155 mL dichloromethane, filtered and again evaporated to give 30.5 g crude product as a slightly brownish foam. This material was dissolved in 122 mL methanol, resulting in a thick suspension, which dissolved on heating to reflux. After 20 min of reflux the solution was allowed to gradually cool to RT during 2 h, whereas crystallization started after 10 min. After 2 h the suspension was cooled to 0° C. for 1 h, followed by −25° C. for 1 h. The crystals were filtered off via a pre-cooled glass sinter funnel, washed portionwise with 78 mL TBME and dried for 18 h at 45° C./20 mbar, to give 21.0 g of the title product as white crystals (77% yield; assay: 99.5%).
MS: m/e 478 (M+H)+, 437, 422.
e) Preparation of (2S,3S,11bS)-1-(2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4(S)-fluoromethyl-pyrrolidin-2-one dihydrochlorideA 2.5 L reactor equipped with a mechanical stirrer, a Pt-100 thermometer, a dropping funnel and a nitrogen inlet was charged with 619 g (1.30 mol) of (2S,3S,11bS)-3-((4S)-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester, 4.2 L isopropanol and 62 mL water and the suspension was heated to 40-45° C. In a second vessel, 1.98 L isopropanol was cooled to 0° C. and 461 mL (6.50 mol) acetyl chloride was added during 35 min, maintaining the temperature at 0-7° C. After completed addition, the mixture was allowed to reach ca. 15° C. and was then slowly added to the first vessel during 1.5 h. After completed addition the mixture was stirred for 18 h at 40-45° C., whereas crystallization started after 1 h. The white suspension was cooled to 20° C. during 2 h, stirred at that temperature for 1.5 h and filtered. The crystals were washed portionwise with 1.1 L isopropanol and dried for 72 h at 45° C./20 mbar, to give 583 g of the product as white crystals (100% yield; assay: 99.0%).
These compounds are useful intermediates for the preparation of DPP-IV inhibitors as disclosed in PCT International Patent Appl. WO 2005/000848. More preferably, the invention relates to a process for the preparation of (2S,3S,11bS)-(3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)]-carbamic acid tert-butyl ester.
XXXXXXX
According to still another embodiment (Scheme 2, below) the (S)-4-fluoromethyl-dihydro-furan-2-one (VII) is directly coupled with the amino-pyrido[2,1-a]isoquinoline derivative (VI) to form the hydroxymethyl derivative of the pyrido[2,1-a]isoquinoline (VIII), which is then subsequently cyclized to the fluoromethyl-pyrrolidin-2-one derivative (IX). The latter can be deprotected to yield the desired pyrido[2,1-a]isoquinoline derivative (I).

In a further preferable embodiment, the process for the preparation of (S)-1-((2S,3S,11bS)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one or of a pharmaceutically acceptable salt thereof comprises the subsequent steps:
- e) coupling of the (2S,3S,11bS)-3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester (amine of formula VI, wherein R2 and R3 are methoxy, R4 is hydrogen and Prot is Boc) with the (S)-4-fluoromethyl-dihydro-furan-2-one of formula
![Figure US20080071087A1-20080320-C00026]()
- f) cyclization of the obtained (2S,3S,11bS)-3-(3-fluoromethyl-4-hydroxy-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester in the presence of a base, and
- g) deprotecting the obtained (2S,3S,11bS)-3-((4S)-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester.

………………………………………………………….
PATENT
http://www.google.com.ar/patents/US7122555?cl=pt-PT
Example 23
RACEMIC
1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one

a) 4-Fluoromethyl-dihydro-furan-2-one
A solution of 4-hydroxymethyl-dihydro-furan-2-one (Tetrahedron 1994, 50, 6839; 1.02 g, 8.78 mmol) and bis(2-methoxyethyl)aminosulfur trifluoride (3.88 g, 17.6 mmol) in chloroform (4.4 mL) was stirred at 40° C. for 1 h, then poured onto ice and partitioned between sat. aq. sodium hydrogencarbonate solution and dichloromethane. The organic layer was washed with brine, dried (MgSO4), and evaporated. Chromatography (SiO2, heptane-ethyl acetate gradient) afforded the title compound (576 mg, 56%). Colourless liquid, MS (EI) 118.9 (M+H)+.
b) 3-Chloromethyl-4-fluoro-butyryl chloride
A mixture of 4-fluoromethyl-dihydro-furan-2-one (871 mg, 7.37 mmol), thionyl chloride (4.39 g, 36.9 mmol), and zinc chloride (60 mg, 0.44 mmol) was stirred 72 h at 80° C., then excess thionyl chloride was removed by distillation. Kugelrohr distillation of the residue (85° C., 0.2 mbar) afforded the title compound (450 mg, 35%). Colourless liquid, 1H-NMR (300 MHz, CDCl3): 4.65–4.55 (m, 1H), 4.50–4.40 (m, 1H), 3.70–3.60 (m, 2H), 3.25–3.05 (m, 2H), 2.80–2.60 (m, 1H).
c) (RS,RS,RS)-[3-(3-Chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
The title compound was produced in accordance with the general method of Example 5c from (RS,RS,RS)-(3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester (Example 5b) and 3-chloromethyl-4-fluoro-butyryl chloride. White solid, MS (ISP) 514.5 (M+H)+.
d) (RS,RS,RS)-[3-(4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
The title compound was produced in accordance with the general method of Example 5d from (RS,RS,RS)-[3-(3-chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester. Off-white foam, MS (ISP) 478.5 (M+H)+.
e) 1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one
The title compound was produced in accordance with the general method of Example 1e from (RS,RS,RS)-[3-(4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester. Light yellow oil, MS (ISP) 378.5 (M+H)+.
Examples 28 and 29
(SR)-1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one
 UNDESIRED
UNDESIREDand
(RS,RS,RS,RS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one

The title compounds were produced from 1-((RS,RS,RS)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one (Example 23) by chromatographic separation (SiO2, CH2Cl2/MeOH/NH4OH 80:1:0.2, then 95:5:0.25).
(SR)-1-((RS,RS,RS)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one: Yellow oil, Rf=0.45 (CH2Cl2/MeOH/NH4OH 90:10:0.25).
(RS,RS,RS,RS)-1-(2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one: Light yellow solid, Rf=0.40 (CH2Cl2/MeOH/NH4OH 90:10:0.25).
Example 30
(S)-1-((S,S,S)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one Dihydrochloride
 DESIRED
DESIREDa) [(S,S,S)-3-(3-Chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
The title compound was produced in accordance with the general method of Example 5c from (S,S,S)-(3-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl)-carbamic acid tert-butyl ester (Example 16b) and 3-chloromethyl-4-fluoro-butyryl chloride (Example 23b). Off-white solid.
b) [(S,S,S)-3-((S)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester and [(S,S,S)-3-((R)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester
Sodium hydride (55–65% dispersion in oil, 1.14 g, 28.5 mmol) was added to a suspension of [(S,S,S)-3-(3-chloromethyl-4-fluoro-butyrylamino)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (6.72 g, 13.1 mmol) in N,N-dimethylformamide (95 mL) at r.t., then after 1 h the reaction mixture was poured onto ice and partitioned between ethyl acetate and water. The organic layer was washed with brine, dried (MgSO4), and evaporated. Chromatography (SiO2, cyclohexane/2-propanol 4:1) afforded [(S,S,S)-3-((S)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (2.40 g, 38%) and the epimer, [(S,S,S)-3-((R)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (2.73 g, 44%).
[(S,S,S)-3-((S)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester: Light yellow foam, Rf=0.6 (SiO2, cyclohexane/2-propanol 1:1).
[(S,S,S)-3-((R)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester: Light yellow foam, Rf=0.4 (SiO2, cyclohexane/2-propanol 1:1).
-
- c) (S)-1-((S,S,S)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido[2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one dihydrochloride
[(S,S,S)-3-((S)-4-Fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (2.40 g, 5.02 mmol) was converted to (S)-1-((S,S,S)-2-amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one in accordance with the general method of Example 1e. The product was dissolved in 2-propanol (10 mL) and treated with hydrogen chloride (5–6 M in 2-propanol, 37 mL). The suspension formed was stirred for 64 h at r.t., then the precipitate was collected by filtration and dried, to afford the title compound (2.04 g, 91%). White solid, m.p. >300° C.
Example 31(R)-1-((S,S,S)-2-Amino-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-3-yl)-4-fluoromethyl-pyrrolidin-2-one dihydrochloride
 UNDESIRED
UNDESIREDThe title compound was produced in accordance with the general method of Example 30c from [(S,S,S)-3-((R)-4-fluoromethyl-2-oxo-pyrrolidin-1-yl)-9,10-dimethoxy-1,3,4,6,7,11b-hexahydro-2H-pyrido [2,1-a]isoquinolin-2-yl]-carbamic acid tert-butyl ester (Example 30b). White solid, m.p. >300° C.

DR ANTHONY MELVIN CRASTO

MY BLOGS ON MED CHEM

DR ANTHONY MELVIN CRASTO Ph.D
web link

New Drug Approvals, ALL ABOUT DRUGS, WORLD DRUG TRACKER
MEDICINAL CHEM INTERNATIONAL, DRUG SYN INTERNATIONAL
SCALEUP OF DRUGS, ALL FOR DRUGS ON WEB,
MY CHINA, VIETNAM AND JAPAN BLOGS
ICELAND, RUSSIA, ARAB
BOBRDOBR, BLAND ICELAND, 100zakladok, adfty
GROUPS
you can post articles and will be administered by me on the google group which is very popular across the world
OPD GROUPSPACES, SCOOP OCI, organic-process-development GOOGLE, TVINX, MENDELEY WDT, SCIPEOPLE OPD, EPERNICUS OPD, SYNTHETIC ORGANIC CHEMISTRYLinkedIn group, DIIGO OPD, LINKEDIN OPD, WDT LINKEDIN, WDTI ZING

Filed under: DIABETES, Phase2 drugs Tagged: CARMEGLIPTIN, DIABETES, phase 2, Roche







