Estrogen Related Receptor alpha (ERR-a) modulators useful for treating, ameliorating, or inhibiting the progression of disease states, disorders, and
conditions mediated by ERR-a activity. BACKGROUND OF THE INVENTION
Nuclear receptors are members of a superfamily of transcription factors.
The members of this family share structural similarities and regulate a diverse set of biological effects (Olefsky, J. M. J. Biol. Chem. 2001 , 276(40), 36863-36864). Ligands activate or repress these transcription factors that control genes involved in metabolism, differentiation and reproduction (Laudet, V. and H. Gronmeyer. The Nuclear Receptor Factbooks. 2002, San Diego: Academic Press). Presently, the human genome project has identified about 48 members for this family and cognate ligands have been identified for about 28 of them (Giguere, V. Endocrine Rev. 1999, 20(5), 689-725). This protein family is composed of modular structural domains that can be interchanged within the members of the family without loss of function. A typical nuclear receptor contains a hypervariable N-terminus, a conserved DNA binding domain (DBD), a hinge region, and a conserved ligand- binding domain (LBD). The function of the DBD is targeting of the receptor to specific DNA sequences (Nuclear Hormone Receptor (NHR) response elements or NREs), and the function of the LBD is recognition of its cognate ligand. Within the sequence of the nuclear receptor there are regions involved in transcriptional activation. The Activation Function 1 (AF-1 ) domain is situated at the N-terminus and constitutively activates transcription (Rochette-Egly, C. et al. Cell 1997, 90, 97-107; Rochette-Egly, C. et al. Mol. Endocrinol. 1992, 6, 2197-2209), while the Activation Function 2 (AF-2) domain is embedded within the LBD and its transcriptional activation is ligand dependent (Wurtz, J.M. et al. Nat. Struct. Biol. 1996, 3, 87-94). Nuclear receptors can exist as monomers, homodimers or heterodimers and bind to direct or inverted nucleotide repeats (Laudet and
Gronmeyer, 2002; Aranda, A. and A. Pascual. Physiol. Rev. 2001 , 81 (3), 1269- 1304).
The members of this family exist either in an activated or repressed basal biological state. The basic mechanism of gene activation involves ligand dependent exchange of co-regulatory proteins. These co-regulatory proteins are referred to as co-activators or co-repressors (McKenna, L.J. et al. Endocrine Rev. 1999, 20, 321 -344). A nuclear receptor in the repressed state is bound to its DNA response element and is associated with co-repressor proteins that recruit histone de-acetylases (HDACs) (Jones, P.L. and Y.B. Shi. Curr. Top. Microbiol. Immunol. 2003, 274, 237-268). In the presence of an agonist there is an exchange of co- repressors with co-activators that in turn recruit transcription factors that assemble into an ATP dependent chromatin-remodeling complex. Histones are hyper- acetylated, causing the nucleosome to unfold, and repression is alleviated. The AF-2 domain acts as the ligand dependent molecular switch for the exchange of co-regulatory proteins. In the presence of an agonist the AF-2 domain undergoes a conformational transition and presents a surface on the LBD for interaction with co-activator proteins. In the absence of an agonist or in the presence of an antagonist the AF-2 domain presents a surface that promotes interactions with co- repressor proteins. The interaction surfaces on the LBD for both co-activators, and co-repressors overlap and provide a conserved molecular mechanism for gene activation or repression that is shared by the members of this family of transcription factors (Xu, H.E. et al. Nature 2002, 415 (6873), 813-817).
Natural ligands that modulate the biological activity of nuclear receptors have been identified for only approximately one half of known nuclear receptors. Receptors for which no natural ligand has been identified are termed “orphan receptors.” The discovery of ligands or compounds that interact with an orphan receptor will accelerate the understanding of the role of the nuclear receptors in physiology and disease and facilitate the pursuit of new therapeutic approaches. Estrogen related receptors (ERRs) constitutes a sub-class of these receptors where no ligand has been identified.
ERR-a (also known as ERR-1 ), an orphan receptor, is the first of the three identified members of the estrogen receptor related subfamily of orphan nuclear receptors (ERR-a, β, γ). The ERR subfamily is closely related to the estrogen receptors (ER-a and ER-β). ERR-a and ERR-β were first isolated by a low stringency hybridization screen (Giguere, V. et al. Nature 1988, 331 , 91 -94) followed later with the discovery of ERR-γ (Hong, H. et al. J. Biol. Chem. 1999, 274, 22618-22626). The ERRs and ERs share sequence similarity with the highest homology observed in their DBDs, approximately 60%, and all interact with the classical DNA estrogen response element. Recent biochemical evidence suggested that the ERRs and ERs share target genes, including pS2, lactoferin, aromatase and osteopontin, and share co-regulator proteins (Giguere, V. Trends in Endocrinol. Metab. 2002, 13, 220-225; Vanacker, J.M. et al. EMBO J. 1999, 18, 4270-4279; Kraus, R.J. et al. J. Biol. Chem. 2002, 272, 24286-24834; Hong et al., 1999; Zhang, Z. and C.T. Teng. J. Biol. Chem. 2000, 275, 20387-20846).
Therefore, one of the main functions of ERR is to regulate the response of estrogen responsive genes. The effect of the steroid hormone estrogen is primarily mediated in the breast, bone and endometrium. Thus, the identification of compounds that will interact with ERRs should provide a benefit for the treatment of bone related disease, breast cancer and reproduction.
ERR-a is shown to be present both in normal and breast cancer tissue (Ariazi, E.A. et al. Cancer Res. 2002, 62, 6510-6518). It has been reported that the main function of ERR-a in normal breast tissue is that of a repressor for estrogen responsive genes. In breast cancers or cell lines that are non-estrogen responsive (ER-a negative), ERR-a has been reported to be in an activated state (Ariazi et al., 2002). Therefore, compounds that will interact with ERR-a may be useful agents for the treatment of breast cancer that is ER-a negative and non- responsive to classical anti-estrogenic therapy, or may be used as an adjunct agent for anti-estrogen responsive breast cancers. These agents may act as antagonists by reducing the biological activity of ERR-a in these particular tissues.
Many post-menopausal women experience osteoporosis, a condition that is a result of the reduction of estrogen production. Reduction of estrogen levels results in an increase of bone loss (Turner, R.T. et al. Endocrine Rev. 1994, 15(3), 275-300). An anabolic effect on bone development has been observed on the administration of estrogens to postmenopausal patients with osteoporosis (Pacifici, R. J. Bone Miner. Res. 1996, 1 1 (8), 1043-1051 ) but the molecular mechanism is unknown since ER-a and ER-β knock-out animals have minor skeletal defects, where the action of estrogens is typically mediated (Korach, K. S. Science 1994, 266, 1524-1527; Windahl, S.H. et al. J. Clin. Invest. 1999, 104(7), 895-901 ). Expression of ERR-a in bone is regulated by estrogen (Bonnelye, E. et al. Mol. Endocrin. 1997, 1 1 , 905-916; Bonnelye, E. et al. J. Cell Biol. 2001 , 153, 971 -984). ERR-a is maintained throughout osteoblast differentiation stages.
Over-expression of ERR-a in rat calvaria osteoblasts, an accepted model of bone differentiation, results in an increase of bone nodule formation, while treatment of rat calvaria osteoblasts with ERR-a antisense results in a decrease of bone nodule formation. ERR-a also regulates osteopontin, a protein believed to be involved in bone matrix formation. Therefore compounds that will modulate ERR-a by increasing its activity can have an anabolic effect for the regeneration of bone density and provide a benefit over current approaches that prevent bone loss, but have no anabolic effect. Such compounds can enhance the activity of the receptor by two possible mechanisms: i) enhancing the association of the receptor with proteins that enhance its activity or improve the stability of the receptor; and ii) increasing the intracellular concentrations of the receptor and consequently increasing its activity. Conversely, with respect to bone diseases that are a result of abnormal bone growth, compounds that will interact with ERR-a and decrease its biological activity may provide a benefit for the treatment of these diseases by retarding bone growth. Antagonism of the association of the receptor with co- activator proteins decreases the activity of the receptor.
ERR-a is also present in cardiac, adipose, and muscle tissue and forms a transcriptional active complex with the PGC-1 co-activator family, co-activators implicated with energy homeostasis, mitochondria biogenesis, hepatic
gluconeogenesis and in the regulation of genes involved in fatty acid beta- oxidation (Kamei, Y. et al. Proc. Natl. Acad. Sci. USA 2003, 100(21 ), 12378- 12383). ERR-a regulates the expression of the medium chain acyl-CoA
dehydrogenase promoter (MCAD). Medium chain acyl-CoA dehydrogenase is a gene involved in the initial reaction in fatty acid beta-oxidation. It is believed that in the adipose tissue ERR-a regulates energy expenditure through the regulation of MCAD (Sladek, R. et al. Mol. Cell. Biol. 1997, 17, 5400-5409; Vega, R.B. and D.P. Kelly. J. Biol. Chem. 1997, 272, 31693-31699). In antisense experiments in rat calvaria osteoblasts, in addition to the inhibition of bone nodule formation, there was an increase in adipocyte differentiation markers including aP2 and PPAR-γ (Bonnelye, E. et al. Endocrinology 2002, 143, 3658-3670). Recently an ERR-a knockout model has been described that exhibited reduced fat mass relative to the wild type and DNA chip analysis data indicated alteration of the expression levels of genes involved in adipogenesis and energy metabolism (Luo, J. et al. Mol. Cell. Biol. 2003, 23(22), 7947-7956). More recently it has been shown that ERR-a regulates the expression of endothelial nitric oxide synthase, a gene that has a protective mechanism against arteriosclerosis (Sumi, D. and L.J. Ignarro. Proc Natl. Acad. Sci. 2003, 100, 14451 -14456). The biochemical evidence supports the involvement of ERR-a in metabolic homeostasis and differentiation of cells into adipocytes. Therefore, compounds interacting with ERR-a can affect energy homeostasis and may therefore provide a benefit for the treatment of obesity and metabolic syndrome related disease indications, including arteriosclerosis and diabetes (Grundy, S.M. et al. Circulation 2004, 109(3), 433-438).
There is a continuing need for new ERR-a inverse agonists. There is also a need for ERR-a inverse agonists useful for the treatment of conditions including but not limited to ankylosing spondylitis, artherosclerosis, arthritis (such as rheumatoid arthritis, infectious arthritis, childhood arthritis, psoriatic arthritis, reactive arthritis), bone-related diseases (including those related to bone formation), breast cancer (including those unresponsive to anti-estrogen therapy), cardiovascular disorders, cartilage-related disease (such as cartilage injury/loss, cartilage degeneration, and those related to cartilage formation),
chondrodysplasia, chondrosarcoma, chronic back injury, chronic bronchitis, chronic inflammatory airway disease, chronic obstructive pulmonary disease, diabetes, disorders of energy homeostasis, gout, pseudogout, lipid disorders, metabolic syndrome, multiple myeloma, obesity, osteoarthritis, osteogenesis imperfecta, osteolytic bone metastasis, osteomalacia, osteoporosis, Paget’s disease, periodontal disease, polymyalgia rheumatica, Reiter’s syndrome, repetitive stress injury, hyperglycemia, elevated blood glucose level, and insulin resistance.
Scheme 1



Scheme 2


Scheme 3


Scheme 4


Scheme 5



Scheme 6

Scheme 7

Scheme 8

Scheme 9

without methyl
Example 199
(5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-3-(c/‘s- 4-fluoropiperidin-3-yl)-1 ,3-thiazolidine-2,4-dione
 note………..this is without methyl
note………..this is without methyl(A) 1 ,1 -Dimethylethyl c/‘s-3-[(5Z)-5-[(1 -[4-chloro-2-(trifluoromethyl)benzyl]-1 H- indazol-5-yl)methylidene]-2,4-dioxo-1 ,3-thiazolidin-3-yl]-4-fluoropiperidine- 1 -carboxylate was prepared from (5Z)-5-({1 -[2-chloro-4-
(trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-2,4-dioxo-1 ,3- thiazolidine (from Example 1 ) and 1 ,1 -dimethylethyl frans-3-hydroxy-4- fluoropiperidine-1 -carboxylate (prepared as described in US 2007/249589) following General Procedure W.
(B) (5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5- yljmethylidene)- 3-(c/s-4-fluoropiperidin-3-yl)-1 ,3-thiazolidine-2,4-dione was prepared from 1 ,1 -dimethylethyl c/s-3-[(5Z)-5-[(1 -[4-chloro-2- (trifluoromethyl)benzyl]-1 /-/-indazol-5-yl)methylidene]-2,4-dioxo-1 ,3- thiazolidin-3-yl]-4-fluoropiperidine-1 -carboxylate following General
Procedure M.
1 H NMR (400 MHz, CDCI3): δ 8.21 (s, 1 H), 7.95 (s, 1 H), 7.72 (d, 1 H), 7.65 (s, 1 H), 7.45 – 7.50 (m, 1 H), 7.30 – 7.38 (m, 2H), 6.66 (d, 1 H), 5.80 (s, 2H), 4.83 – 5.04 (m, 2H), 4.08 – 4.20 (m, 2H), 3.99 – 4.08 (m, 1 H), 3.81 – 3.91 (m, 1 H), 2.27 – 2.40 (m, 1 H), 2.02 – 2.13 (m, 1 H).
LC/MS: mass calcd. for C24Hi9CIF4N4O2S: 538.08, found 539.5 [M+1 ]+
Example 201
(5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-3-(c/‘s- 3-fluoropiperidin-4-yl)-1 ,3-thiazolidine-2,4-dione
 note. this is without methyl
note. this is without methyl(A) 1 ,1 -Dimethylethyl c/‘s-4-[(5Z)-5-[(1 -[4-chloro-2-(trifluoromethyl)benzyl]-1 H- indazol-5-yl)methylidene]-2,4-dioxo-1 ,3-thiazolidin-3-yl]-3-fluoropiperidine- 1 -carboxylate was prepared from (5Z)-5-({1 -[2-chloro-4- (trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-2,4-dioxo-1 ,3- thiazolidine (from Example 1 ) and 1 ,1 -dimethylethyl frans-4-hydroxy-3- fluoropiperidine-1 -carboxylate (prepared as described in US 2007/249589) following General Procedure J.(B) (5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5- yl}methylidene)-3-(c/s-3-fluoropiperidin-4-yl)-1 ,3-thiazolidine-2,4-dione was prepared from 1 ,1 -dimethylethyl c/‘s-4-[(5Z)-5-[(1 -[4-chloro-2- (trifluoromethyl)benzyl]-1 /-/-indazol-5-yl)methylidene]-2,4-dioxo-1 ,3- thiazolidin-3-yl]-3-fluoropiperidine-1 -carboxylate following General
Procedure M.
1 H NMR (400 MHz, CDCI3): δ 8.22 (s, 1 H), 8.00 (s, 1 H), 7.96 (s, 1 H), 7.72 (d, 1 H), 7.48 – 7.54 (m, 1 H), 7.36 (s, 1 H), 7.34 (s, 1 H), 6.68 (d, 1 H), 5.80 (s, 2H), 4.57 – 4.75 (m, 1 H), 4.40 – 4.56 (m, 1 H), 3.25 – 3.46 (m, 2H), 3.18 (qd, 1 H), 2.83 – 3.03 (m, 1 H), 2.72 (t, 1 H), 1 .88 (br. s., 1 H), 1 .72 (d, 1 H).
LC/MS: mass calcd. for C2 H19CIF4N4O2S: 538.08, found 539.5 [M+1 ]+
Example 273
(5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-3- (frans-3-fluoropiperidin-4-yl)-1 ,3-thiazolidine-2,4-dione
 note——- this is without methyl but precursor to desired compd
note——- this is without methyl but precursor to desired compdPreparation 1 :
(A) To the solution of 1 ,1 -dimethylethyl frans-4-(2,4-dioxo-1 ,3-thiazolidin-3-yl)- 3-hydroxypiperidine-1 -carboxylate (from Example 270, 0.68 mmol) in DCM (5 ml_) in a plastic bottle was added bis(2-methoxyethyl)aminosulfur trifluoride (3 equiv) and a drop of ethanol. After stirring at rt for 3 h, the reaction was concentrated and the resultant residue was purified by silica gel chromatography (hexane/EtOAc) to provide 1 ,1 -dimethylethyl trans-4- (2,4-dioxo-1 ,3-thiazolidin-3-yl)-3-fluoropiperidine-1 -carboxylate as a pale yellow solid.
(B) (5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5-yl}methylidene)- 3-[frans-3-fluoropiperidin-4-yl]-1 ,3-thiazolidine-2,4-dione was prepared from [4-chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5-carbaldehyde (from
Example 1 ) and 1 ,1 -dimethylethyl frans-4-(2,4-dioxo-1 ,3-thiazolidin-3-yl)-3- fluoropiperidine-1 -carboxylate following General Procedure F.
Preparation 2:
(A) A mixture of 1 ,1 -dimethylethyl 7-oxa-3-azabicyclo[4.1 .0]heptane-3- carboxylate (from Example 270; 47.7 mmol), [(5Z)-5-({1 -[4-chloro-2- (trifluoromethyl)benzyl]-1 /-/-indazol-5-yl}methylidene)-2,4-dioxo-1 ,3- thiazolidine (from Example 1 ; 31 .8 mmol) and magnesium perchlorate (23.9 mmol) in DMF (70 mL) was heated at 1 15 °C for 2-4 h. After cooling to rt, the mixture was slowly poured into water (300 mL) with vigorous stirring, and the resultant precipitate was filtered, thoroughly washed with water and dried to afford a mixture of 1 ,1 -dimethylethyl frans-4-{(5Z)-5-[(1 -
{[4-chloro-2-(trifluoromethyl)phenyl]methyl}-1 /-/-indazol-5-yl)methylidene]- 2,4-dioxo-1 ,3-thiazolidin-3-yl}-3-hydroxypiperidine-1 -carboxylate and the corresponding regioisomer, 1 ,1 -dimethylethyl frans-3-{(5Z)-5-[(1 -{[4-chloro- 2-(trifluoromethyl)phenyl]methyl}-1 /-/-indazol-5-yl)methylidene]-2,4-dioxo- 1 ,3-thiazolidin-3-yl}-4-hydroxypiperidine-1 -carboxylate in ratio of ~ 3.3 : 1 .
(B) To an ice-cooled solution of the above mixture of 1 ,1 -dimethylethyl frans- 4-{(5Z)-5-[(1 -{[4-chloro-2-(trifluoromethyl)phenyl]methyl}-1 /-/-indazol-5- yl)methylidene]-2,4-dioxo-1 ,3-thiazolidin-3-yl}-3-hydroxypiperidine-1 – carboxylate and the regioisomer, 1 ,1 -dimethylethyl frans-3-{(5Z)-5-[(1 -{[4- chloro-2-(trifluoromethyl)phenyl]methyl}-1 H-indazol-5-yl)methylidene]-2,4- dioxo-1 ,3-thiazolidin-3-yl}-4-hydroxypiperidine-1 -carboxylate in DCM (350 mL) was slowly added bis(2-methoxyethyl)aminosulfur trifluoride (47.7 mmol). After stirring for 1 h, the solution was allowed to warm to rt and stir overnight. The reaction was then quenched with sat’d aq. NaHCO3 and after separating phases, the organic phase was dried (Na2SO4) and concentrated to ~ 40 mL. The solution was loaded onto a silica gel column (Analogix, 200g) and eluted with heptanes/DCM/EtOAc (40:57:3).
Product-containing fractions were combined and concentrated to afford a crude product mixture as a pale yellow foam. Treatment of this foam with ether (~ 20 mL) led to product precipitation; additional ether (200 mL) was added portionwise with stirring and after cooling to ~ 5 °C, the mixture was filtered through a glass fiber filter and washed with cold ether to afford 1 ,1 – dimethylethyl frans-4-{(5Z)-5-[(1 -{[4-chloro-2-(trifluoromethyl)phenyl]- methyl}-1 H-indazol-5-yl)methylidene]-2,4-dioxo-1 ,3-thiazolidin-3-yl}-3- fluoropiperidine-1 -carboxylate as an essentially white powder. (C) (5Z)-5-({1 -[4-Chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5- yl}methylidene)-3-[frans-3-fluoropiperidin-4-yl]-1 ,3-thiazolidine-2,4-dione was prepared from 1 ,1 -dimethylethyl frans-4-{(5Z)-5-[(1 -{[4-chloro-2- (trifluoromethyl)phenyl]methyl}-1 H-indazol-5-yl)methylidene]-2,4-dioxo-1 ,3- thiazolidin-3-yl}-3-fluoropiperidine-1 -carboxylate following General
Procedure M.
1 H NMR (400 MHz, CDCI3): δ 8.22 (s, 1 H), 8.02 (s, 1 H), 7.96 (s, 1 H), 7.72 (d, 1 H), 7.47 – 7.56 (m, 1 H), 7.36 (s, 1 H), 7.34 (s, 1 H), 6.68 (d, 1 H), 5.80 (s, 2H), 5.10 – 5.33 (m, 1 H), 4.40 – 4.55 (m, 1 H), 3.52 (d, 1 H), 3.14 (d, 1 H), 2.68 (br. s., 2H), 2.43 (qd, 1 H), 1 .70 – 1 .90 (m, 2H).
LC/MS: mass calcd. for C2 H2oCIF4N4O2S: 538.09, found 539.3 [M+1 ]+
main compd
Example 277
(5Z)-5-({1-[4-Chloro-2-(trifluoromethyl)benzyl]-1H-indazol-5-yl}methylidene)-3- (frans-3-fluoro-1-methylpiperidin-4-yl)-1 ,3-thiazolidine-2,4-dione
desired compd(5Z)-5-({1-[4-Chloro-2-(trifluoromethyl)benzyl]-1H-indazol-5-yl}methylidene)- 3-[ trans -3-fluoro-1-methylpiperidin-4-yl]-1,3-thiazolidine-2,4-dione was prepared from (5Z)-5-({1 -[4-chloro-2-(trifluoromethyl)benzyl]-1 H-indazol-5- yl}methylidene)-3-[ trans -3-fluoropiperidin-4-yl]-1 ,3-thiazolidine-2,4-dione (Example 273) and formaldehyde following General Procedure R.
1 H NMR (400 MHz, CDCI3): δ 8.22 (s, 1 H), 8.01 (s, 1 H), 7.96 (s, 1 H), 7.72 (s, 1 H), 7.51 (d, 1 H), 7.36 (s, 1 H), 7.34 (s, 1 H), 6.68 (d, 1 H), 5.80 (s, 2H), 5.25 – 5.48 (m, 1 H), 4.28 – 4.42 (m, 1 H), 3.24 – 3.36 (m, 1 H), 2.85 – 2.96 (m,
1 H), 2.56 (qd, 1 H), 2.37 (s, 3H), 2.07 – 2.17 (m, 2H), 1 .77 (dd, 1 H).
LC/MS: mass calcd. for C25H2iCIF4N4O2S: 552.1 , found 553.3 [M+1 ]+

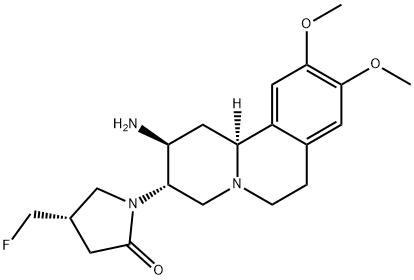
The development of a reproducible process for multihundred gram production of (Z)-5-((1-(4-chloro-2-(trifluoromethyl)benzyl)-1H-indazol-5-yl)methylene)-3-((3R,4R)-3-fluoro-1-methylpiperidin-4-yl)thiazolidine-2,4-dione (26), a potent and selective inhibitor of estrogen-related receptor 1 (ERR1), is described. This multihundred gram synthesis was achieved via magnesium perchlorate-catalyzed regioselective epoxide ring-opening of tert-butyl 7-oxa-3-azabicyclo[4.1.0]heptane-3-carboxylate (9) with thiazolidine-2,4-dione (6, TZD) to form a diastereomeric mixture tert-butyl 4-(2,4-dioxothiazolidin-3-yl)-3-hydroxypiperidine-1-carboxylate (17), of which the 3-hydroxyl group was functionally transformed to 3-fluoro derivative 19 after treatment with Deoxo-Fluor. Chiral separation of 19 provided the desired diastereomer (3R,4R)-21 that was converted to the secondary amine 23 TFA salt. Reductive amination of 23 produced the key intermediate N-methyl 24. Knoevenagel condensation of24 with 1-(4-chloro-2-(trifluoromethyl)benzyl)-1H-indazole-5-carbaldehyde (5) produced the final product 26 in 10% overall yield (99.7% HPLC area% with ≥99.5% de) after a convergent eight synthetic steps with the only column purification being the chiral HPLC separation of 3R,4R-21 from 3S,4S-22.
Citations
- Bignan, G; WO 2011149841 2011
- Li, X; 246th American Chemical Society National Meeting 2013
- Slade, D; J Org Chem 2009, 74, 6331
- Collot, V; Tetrahedron 1999, 55, 6917
- Patta, S; Indian J Chem 2005, 2404
- Maccari, R; Bioorg Med Chem 2005, 13, 2809
- Corona, J; Org Process Res Dev 2010, 14, 712
- Chen, S; Bioorg Med Chem Lett 2007, 17, 2134
- Boto, A; Eur J Org Chem 2005, 673
- Saavedra, J; J Org Chem 1979, 44, 4516
- Bosmans, J; WO 2005000838 2005
- Kratzel, M; Heterocycles 1995, 41, 897
- Daly, A; Tetrahedron Lett 1999, 40, 3617
- Cresswell, A; Org Lett 2010, 12, 2936
- Ready, J; Angew Chem, Int Ed 2002, 41, 1394
- Tandon, V; Tetrahedron Lett 1993, 34, 4403
- Zhao, S; Heterocycles 1994, 39, 163
- Imanishi, T; Synth Comm 1978, 8, 99
- White, J; J Org Chem 2004, 69, 2573
- Lal, G; Chem Commun 1999, 215
- Singh, R; Synthesis 2002, 17, 2561
- Singh, R; J Fluorine Chem 2002, 116, 23
- Shaw, S; J Org Chem 2013, 78, 8892
- Grunewald, G; J Med Chem 2001, 44, 2849
Filed under: DIABETES, Uncategorized Tagged: (Z)-5-((1-(4-Chloro-2-(trifluoromethyl)benzyl)-1H-indazol-5-yl)methylene)-3-((3R, 4-dione, 4R)-3-fluoro-1-methylpiperidin-4-yl)thiazolidine-2, DIABETES








 Originally posted on
Originally posted on 
































































 6a-4 is TA 1887
6a-4 is TA 1887
























 R1 = FLUORO, R2= H
R1 = FLUORO, R2= H

















 3p is compd
3p is compd

































![[1860-5397-9-265-7]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-7.png?max-width=550&background=EEEEEE)
![[1860-5397-9-265-i29]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i29.png?max-width=550&background=EEEEEE)
![[1860-5397-9-265-8]](http://www.beilstein-journals.org/bjoc/content/figures/1860-5397-9-265-8.png?max-width=550&background=EEEEEE)
![[1860-5397-9-265-i30]](http://www.beilstein-journals.org/bjoc/content/inline/1860-5397-9-265-i30.png?max-width=550&background=EEEEEE)


































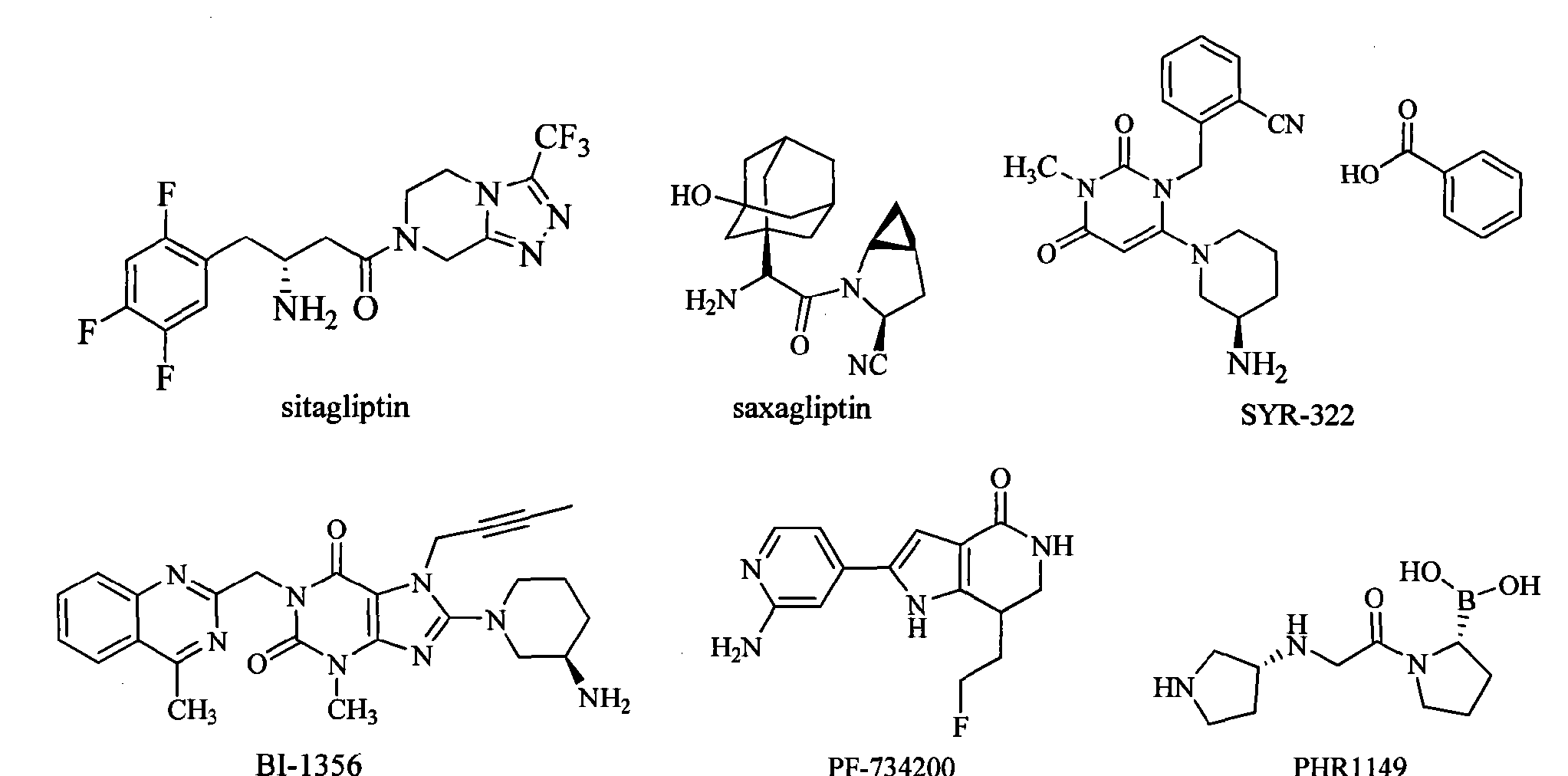
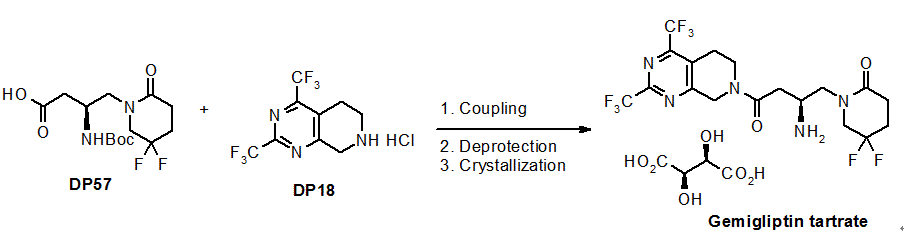
 Nanjing Changao Pharmaceutical 澳格列汀 is a novel DPP-4 inhibitor (gliptin) for the treatment of type II diabetes. Because Shanghai Sun Sail Pharmaceutical, a wholly owned subsidiary of Nanjing Changao Pharmaceutical, has filed two patents to protect DPP-4 inhibitors (WO2011147207 and CN101786978), it is unknown which one covers this drug. Relevant data’s from WHO showed morbidity rate, disability rate, death rate of diabetes mellitus and overall health level of diabetes mellitus patients have already ranked the third place in non-infectious diseases, diabetes, together with tumors and cardiovascular diseases were the three main diseases which threats human health. Diabetes mellitus is usually classified into type 1 and type 2, there are more than 240 million diabetes patients, and 90% of them are suffering from type 2 diabetes, which also has a 1% growth rate every year, so, type 2 diabetes will be the main new growth point of diabetes drug market. The incidence of diabetes in China is about 5%, the number of patients of which ranks second place in the world just behind India. There are many antidiabetic drugs on the market, insulin injection, metformin, rosiglitazone, pioglitazone are representations of them. However, there is no drug alone can keep the HbA1c level of type 2 diabetes patients within the aimed range in a long term. Even though used in combination, the effect of the drugs will go down year by year after 3-4 years. Adverse reaction is one of the problems of many hypoglycemic drugs, wherein the fatal hypoglycemia is most worried by clinicians; secondly, many oral hypoglycemic drugs, such as sulfonylureas, α-glycosidase inhibitors and thiazolidinediones may all induce weight gain to patients, some of the drugs may also induce cardiovascular diseases. Therefore, developing new type hypoglycemic drugs with brand new mechanism of action, higher safety and effectiveness is an important task that should be completed quickly for the scientists. In the process of constantly finding new methods endocrine hormones were found to play an important role in the pathology and physiology of type 2 diabetes. Dipeptidyl peptidase-IV (DPP-IV) is an important enzyme related to diabetes, inhibiting the action of which to treat type 2 diabetes is a new method with good prospect. DPP-IV inhibitors can indirectly stimulate the secretion of insulin, the action of which is generated by inhibit DPP-IV to stabilize endocrine hormones such as incretin hormones, glucagons-like-peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP). GLP-1 is a production expressed by glucagon protogene after eating, and mainly secreted by intestinal mucosa L-cell, and it can stimulate the secretion of insulin by pancreatic β-cells, which plays a significant role in the stability of blood sugar. Experiments prove that GLP-1 has physiological functions as following: acting on pancreatic β-cells in a glucose-dependent manner, facilitating the transcription of insulin genes, increasing the biosynthesis and secretion of insulin, stimulating the proliferation and differentiation of β-cells, inhibiting the apoptosis of β-cells to increasing the number of pancreatic β-cells; inhibiting the secretion of glucagon; inhibiting the appetite and food intake; retarding the emptying of gastric contents, etc., all of these functions are helpful to reduce blood sugar after food intake and to keep blood sugar within constant level. In addition, it won’t cause the danger of severe hypoglycemia. GLP-1 well controlled the blood sugar of type 2 diabetes animal models and patients by multiple mechanisms. However, GLP-1 may lose biological activity through quick degradation by DPP-IV, and the half life of it is shorter than 2 minutes, which utterly limits the clinical use of GLP-1. It was found in researches that DPP-IV inhibitors can totally protect endogenous and even extraneous GLP-1 from inactivation by DPP-IV, improve activated GLP-llevel, and reduce the antagonistic effect of GLP-1 metabolites. Moreover, DPP-IV inhibitors can also delay the incidence of diabetes through stimulating the regeneration of pancreatic β-cells and the improving the glucose tolerance and insulin sensitivity. Dipeptidyl peptidase-IV (DPP-IV) inhibitors represent a novel class of agents that are being developed for the treatment or improvement in glycemic control in patients with Type 2 diabetes. For reviews on the application of DPP-IV inhibitors for the treatment of Type 2 diabetes, reference is made to the following publications: (1)
Nanjing Changao Pharmaceutical 澳格列汀 is a novel DPP-4 inhibitor (gliptin) for the treatment of type II diabetes. Because Shanghai Sun Sail Pharmaceutical, a wholly owned subsidiary of Nanjing Changao Pharmaceutical, has filed two patents to protect DPP-4 inhibitors (WO2011147207 and CN101786978), it is unknown which one covers this drug. Relevant data’s from WHO showed morbidity rate, disability rate, death rate of diabetes mellitus and overall health level of diabetes mellitus patients have already ranked the third place in non-infectious diseases, diabetes, together with tumors and cardiovascular diseases were the three main diseases which threats human health. Diabetes mellitus is usually classified into type 1 and type 2, there are more than 240 million diabetes patients, and 90% of them are suffering from type 2 diabetes, which also has a 1% growth rate every year, so, type 2 diabetes will be the main new growth point of diabetes drug market. The incidence of diabetes in China is about 5%, the number of patients of which ranks second place in the world just behind India. There are many antidiabetic drugs on the market, insulin injection, metformin, rosiglitazone, pioglitazone are representations of them. However, there is no drug alone can keep the HbA1c level of type 2 diabetes patients within the aimed range in a long term. Even though used in combination, the effect of the drugs will go down year by year after 3-4 years. Adverse reaction is one of the problems of many hypoglycemic drugs, wherein the fatal hypoglycemia is most worried by clinicians; secondly, many oral hypoglycemic drugs, such as sulfonylureas, α-glycosidase inhibitors and thiazolidinediones may all induce weight gain to patients, some of the drugs may also induce cardiovascular diseases. Therefore, developing new type hypoglycemic drugs with brand new mechanism of action, higher safety and effectiveness is an important task that should be completed quickly for the scientists. In the process of constantly finding new methods endocrine hormones were found to play an important role in the pathology and physiology of type 2 diabetes. Dipeptidyl peptidase-IV (DPP-IV) is an important enzyme related to diabetes, inhibiting the action of which to treat type 2 diabetes is a new method with good prospect. DPP-IV inhibitors can indirectly stimulate the secretion of insulin, the action of which is generated by inhibit DPP-IV to stabilize endocrine hormones such as incretin hormones, glucagons-like-peptide-1 (GLP-1) and glucose-dependent insulinotropic peptide (GIP). GLP-1 is a production expressed by glucagon protogene after eating, and mainly secreted by intestinal mucosa L-cell, and it can stimulate the secretion of insulin by pancreatic β-cells, which plays a significant role in the stability of blood sugar. Experiments prove that GLP-1 has physiological functions as following: acting on pancreatic β-cells in a glucose-dependent manner, facilitating the transcription of insulin genes, increasing the biosynthesis and secretion of insulin, stimulating the proliferation and differentiation of β-cells, inhibiting the apoptosis of β-cells to increasing the number of pancreatic β-cells; inhibiting the secretion of glucagon; inhibiting the appetite and food intake; retarding the emptying of gastric contents, etc., all of these functions are helpful to reduce blood sugar after food intake and to keep blood sugar within constant level. In addition, it won’t cause the danger of severe hypoglycemia. GLP-1 well controlled the blood sugar of type 2 diabetes animal models and patients by multiple mechanisms. However, GLP-1 may lose biological activity through quick degradation by DPP-IV, and the half life of it is shorter than 2 minutes, which utterly limits the clinical use of GLP-1. It was found in researches that DPP-IV inhibitors can totally protect endogenous and even extraneous GLP-1 from inactivation by DPP-IV, improve activated GLP-llevel, and reduce the antagonistic effect of GLP-1 metabolites. Moreover, DPP-IV inhibitors can also delay the incidence of diabetes through stimulating the regeneration of pancreatic β-cells and the improving the glucose tolerance and insulin sensitivity. Dipeptidyl peptidase-IV (DPP-IV) inhibitors represent a novel class of agents that are being developed for the treatment or improvement in glycemic control in patients with Type 2 diabetes. For reviews on the application of DPP-IV inhibitors for the treatment of Type 2 diabetes, reference is made to the following publications: (1) 
 courtesy yaopha see enlarged image at
courtesy yaopha see enlarged image at 









 DUTOGLIPTIN
DUTOGLIPTIN


















































































































































 LIONEL MY SON
LIONEL MY SON








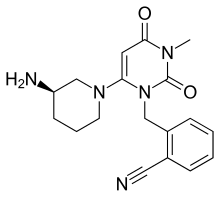

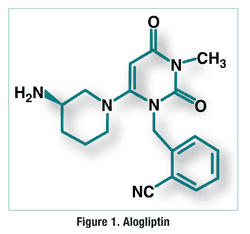




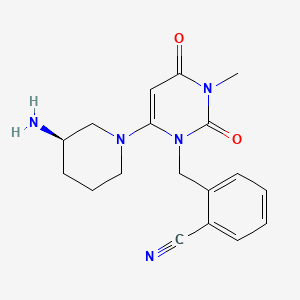


 Alogliptin benzoate
Alogliptin benzoate










%7D)